Freie Universität Berlin
30.04.2020, 11:12 Uhr
Neue Wirkstoffe aus dem Computer
Die Open Source Plattform "VirtualFlow" untersucht Milliarden Moleküle in kürzester Zeit.
(Quelle: Wikimedia (AzaToth) )
Die Entwicklung neuer Wirkstoffe in der Medizin kann viele Jahre dauern und Milliarden Euro kosten. Einem internationalen Team von Wissenschaftlerinnen und Wissenschaftlern mit Beteiligung von Forscherinnen und Forschern der Freien Universität Berlin und der Technischen Universität Berlin ist es jetzt gelungen, eine Plattform zu entwickeln, die Milliarden potenzielle Wirkstoffe in kürzester Zeit untersuchen kann. Die Arbeit wurde in der renommierten Fachzeitschrift Nature publiziert. Angeregt durch die Corona-Pandemie haben die Forscherinnen und Forscher jetzt begonnen, mithilfe dieser Plattform nach potenziellen Wirkstoffen zu suchen, die die Coronavirus-Proteine blockieren könnten.
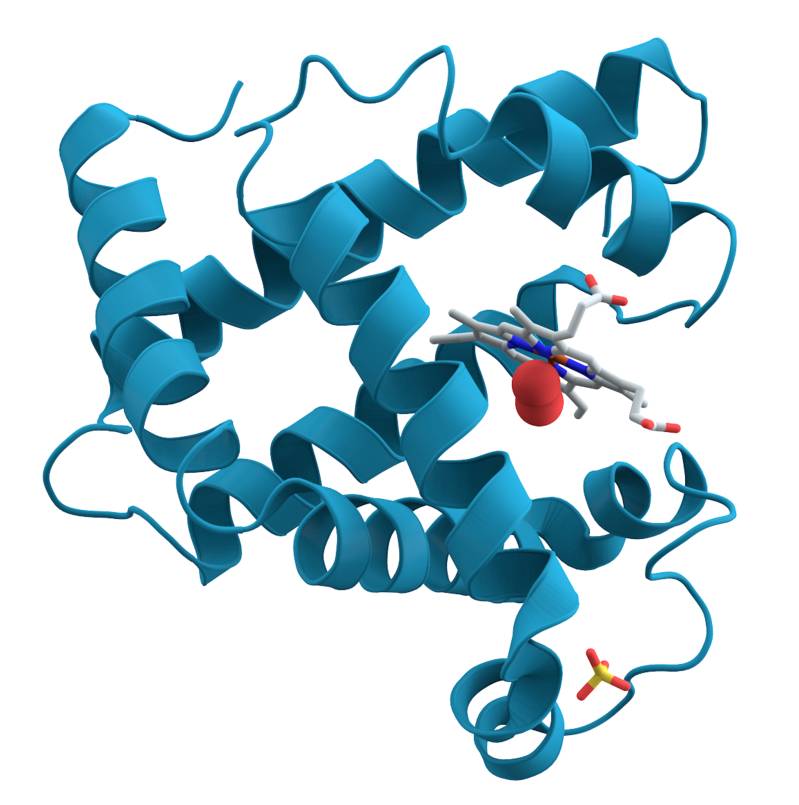
Ein bewährtes Prinzip der medizinischen Wirkstoffforschung ist es, krankmachende Proteine im Körper durch das Einbringen von kleinen Molekülen zu hemmen, die an einer bestimmten Position fest an das krankmachende Protein binden und es so manipulieren oder hemmen. Ein Prinzip, das mit dem Schlüssel-Schloss-Prinzip vergleichbar ist: Die kleinen Moleküle (Schlüssel) passen in eine bestimmte Bindungsstelle (Schloss) des krankmachenden Proteins und schalten es so aus. Das Problem: Es gibt potenziell eine riesige Anzahl an mehr oder weniger gut passenden Schlüsseln, die Grundlage für einen neuen medizinischen Wirkstoff sein könnten und potenziell alle getestet werden müssen. Computersimulationen, die die aufwendigen Laborexperimente ersetzen, haben das Potenzial, diese Probleme zu lösen.
"In einer Computersimulation können wir die Fähigkeit eines kleinen Moleküls, an ein krankmachendes Protein zu binden – auch 'docken' genannt -, testen. Macht man dies mit vielen kleinen Molekülen, nennen wir das virtuelles Screening. Diesen Ansatz gibt es bereits, allerdings konnte bisher routinemäßig nur eine relativ geringe Zahl von kleinen Molekülen in solchen virtuellen Screenings getestet werden", erläutert Christoph Gorgulla, Postdoctoral Research Fellow an der Harvard University, der mit einem Stipendium des Einstein Center for Mathematics (ECMath) an der Freien Universität Berlin promovierte und auf dessen Promotion die wesentlichen Ergebnisse der Veröffentlichung beruhen. "Wir haben nach einer Möglichkeit gesucht, deutlich mehr kleine Moleküle virtuell zu screenen als bisher. Uns interessierte dabei nicht nur die klassische Unterscheidung 'passt' oder 'passt nicht', sondern 'passt besser' und 'passt schlechter'. Denn diese sogenannte Bindungsstärke ist eine der wichtigsten Eigenschaften von Wirkstoffen."
Dazu erstellte das Team eine der größten Molekülbibliotheken der Welt, die Docking-Moleküle bereitstellt, indem es die dreidimensionale Geometrie von mehr als 1,4 Milliarden kleinen Molekülen berechnete und in der Datenbank festhielt. Diese Daten stehen open source zur Verfügung und können von Wirkstoffforscherinnen und Wirkstoffforschern auf der ganzen Welt genutzt werden. Zusätzlich entwickelte das Forschungsteam die Plattform "Virtual Flow", die es ermöglicht, in kurzer Zeit nicht nur Millionen, sondern Milliarden von kleinen Molekülen parallel durch Computersimulationen auf ihre Bindungsfähigkeit an ein bestimmtes Protein zu testen.
"Anstatt auf einem gewöhnlichen Computer läuft 'VirtualFlow' auf Supercomputern, welche viele Tausend Prozessoren enthalten. Es funktioniert auch auf Clouds", so Dr. Konstantin Fackeldey, Privat-Dozent am Institut für Mathematik der TU Berlin, der unter anderem im Rahmen des Berliner Exzellenzclusters Math+ mit dem Zuse-Institut Berlin kooperiert und Teil des Projektteams ist. "Getestet haben wir 'VirtualFlow' zum Beispiel auf der Cloud-Plattform von Google mit bis zu 160.000 Prozessoren." "Virtual Flow" berücksichtigt sogar eine zusätzliche Komplexität: Proteine sind beweglich, sodass sich die spezifische, dreidimensionale Bindungsstelle der kleinen Moleküle an einem Protein unter bestimmten Bedingungen auch verändern kann. Diese potenzielle Beweglichkeit der Bindungsstellen wird in der mathematischen Simulation berücksichtigt.
Google hat den Wissenschaftlerinnen und Wissenschaftlern jetzt zusätzliche Mittel zur Verfügung gestellt, um seine Cloud-Computing-Plattform zu nutzen und damit nach möglichen Kandidaten für Wirkstoffe gegen das Coronavirus zu suchen. "Obwohl virtuelle Screenings sehr schnell sein können, erwarten wir nicht, dass dieser Ansatz kurzfristig zu einem zugelassenen Wirkstoff führt. Aber diese Screenings können die Suche nach potenziellen Wirkstoffen deutlich besser leiten. Letztlich sind für die Zulassung eines Medikamentes aber immer Laborexperimente und klinische Studien notwendig, welche relativ zeitaufwendig sind", betont Konstantin Fackeldey. "VirtualFlow" ist als eine freie Open-Source Plattform verfügbar und steht somit allen Wissenschaftlerinnen und Wissenschaftlern kostenfrei zur Verfügung.
Weitere Informationen: https://www.nature.com/articles/s41586-020-2117-z
Virtual Flow auf GitHub: https://github.com/VirtualFlow











